
解析基因组的“语言”:戈登贝尔奖决赛选手使用大型语言模型来预测新冠病毒变异株
来自美国阿贡国家实验室、NVIDIA、芝加哥大学等组织机构的研究员开发了一个处理基因组规模数据的先进模型,并入围戈登贝尔COVID-19研究特别奖决赛。
这一戈登贝尔特别奖旨在表彰基于高性能计算的COVID-19研究。一位决赛入围选手教会了大型语言模型(LLMs)一种新的语言——基因序列,使这些模型能够提供基因组学、流行病学和蛋白质工程方面的洞察。
这项开创性的成果发表于10月,是由来自美国阿贡国家实验室、NVIDIA、芝加哥大学等组织机构的二十多名学术和商业研究员合作完成。
该研究团队训练了一个LLM来追踪基因突变,并预测需要关注的SARS-CoV-2(导致COVID-19的病毒)变异株。虽然迄今为止大多数应用于生物学的LLM都是在小分子或蛋白质的数据集上训练的,但这一项目是在原始核苷酸序列(DNA和RNA的最小单位)上训练的首批模型之一。
负责带领该项目的阿贡国家实验室计算生物学家Arvind Ramanathan表示:“我们假设从蛋白质水平到基因水平的数据有助于我们构建出更易于理解新冠病毒变异株的模型。通过训练模型去追踪整个基因组及其进化过程中的所有变化,我们不仅能够更好地预测COVID,还能预测已掌握足够基因组数据的任何疾病。”
戈登贝尔奖被誉为HPC领域的诺贝尔奖。今年的戈登贝尔奖将在本周的SC22上由美国计算机协会颁发。该协会代表着全球约10万名计算领域的专家,自2020年开始向使用HPC推进COVID-19研究的杰出研究员颁发特别奖。
在一种只有四个字母的语言上训练大型语言模型
长期以来,LLM一直在接受人类语言的训练,这些语言通常由几十个字母组成,可以排列组合成数万个单词,并连接成长句和段落。而生物学语言只有四个代表核苷酸的字母,即DNA中的A、T、G和C,或RNA中的A、U、G和C。这些字母按不同顺序排列成基因。
虽然较少的字母看似会降低AI学习的难度,但实际上生物学语言模型要复杂得多。这是因为人类的基因组由超过30亿个核苷酸组成,而冠状病毒的基因组由大约3万个核苷酸组成,因此很难将基因组分解成不同、有意义的单位。
Ramanathan表示: “在理解基因组这一 ‘生命代码’的过程中,我们所面对的一个主要挑战是基因组中的庞大测序信息。核苷酸序列的意义可能会受另一序列的影响,以人类的文本做类比,这种影响的范围不仅仅是文本中的下一句话或下一段话,而是相当于一本书中的整个章节。”
参与该项目协作的NVIDIA研究员设计了一种分层扩散方法,使LLM能够将约1500个核苷酸的长字符串当作句子来处理。
论文共同作者、NVIDIA AI研究高级总监、加州理工学院计算+数学科学系布伦讲席教授Anima Anandkumar表示:“标准语言模型难以生成连贯的长序列,也难以学习不同变异株的基本分布。我们开发了一个在更高细节水平上运作的扩散模型,该模型使我们能够生成现实中的变异株,并采集到更完善的统计数据。”
预测需要关注的新冠病毒变异株
该团队首先使用细菌和病毒生物信息学资源中心的开源数据,对来自原核生物(像细菌一样的单细胞生物)超过1.1亿个基因序列进行了LLM预训练,然后使用150万个高质量的新冠病毒基因组序列,对该模型进行微调。
研究员还通过在更广泛的数据集上进行预训练,确保其模型能够在未来的项目中推广到其他预测任务,使其成为首批具备此能力的全基因组规模的模型之一。
在对COVID数据进行了微调后,LLM就能够区分病毒变异株的基因组序列。它还能够生成自己的核苷酸序列,预测COVID基因组的潜在突变,这可以帮助科学家预测未来需要关注的变异株。
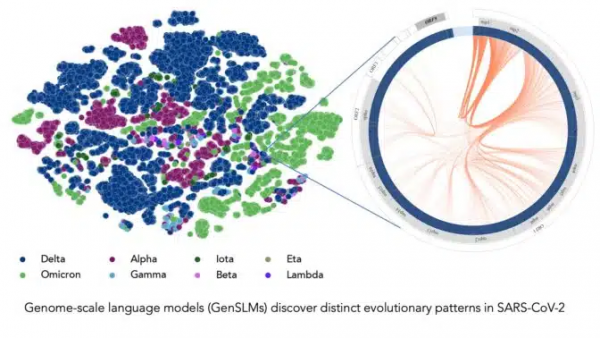
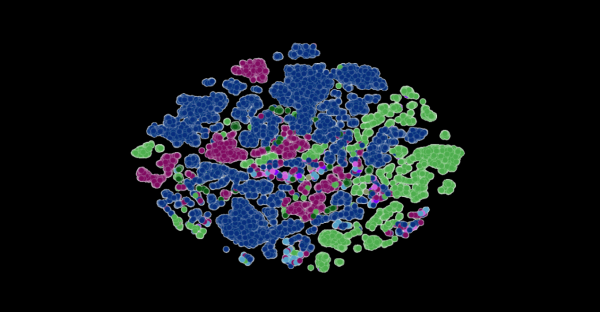
在长达一年时间内积累的SARS-CoV-2基因组数据的训练下,该模型可以推断出各种病毒株之间的区别。左边的每个点对应一个已测序的SARS-CoV-2病毒株,并按变异株颜色编码。右图放大了该病毒的一个特定毒株,它捕捉到了该毒株特有的病毒蛋白进化耦合关系。图片由美国阿贡国家实验室的Bharat Kale、Max Zvyagin和Michael E. Papka提供。
Ramanathan表示: “大多数研究员一直在追踪新冠病毒突刺蛋白的突变,尤其是与人类细胞结合的域。但病毒基因组中还有其他蛋白质也会经历频繁的突变,所以了解这些蛋白质十分重要。”
论文中提到,该模型还可以与AlphaFold、OpenFold等常见的蛋白质结构预测模型整合,帮助研究员模拟病毒结构,研究基因突变如何影响病毒感染其宿主的能力。OpenFold是NVIDIA BioNeMo LLM服务中包含的预训练语言模型之一。NVIDIA BioNeMo LLM服务面向的是致力于将LLM应用于数字生物学和化学应用的开发者。
利用GPU加速超级计算机大幅加快AI训练速度
该团队在由NVIDIA A100 Tensor Core GPU驱动的超级计算机上开发AI模型,包括阿贡国家实验室的Polaris、美国能源部的Perlmutter以及NVIDIA的Selene系统。通过扩展到这些强大的系统,他们在训练中实现了超过1500 exaflops的性能,创建了迄今为止最大的生物语言模型。
Ramanathan 表示: “我们如今处理的模型有多达250亿个参数,预计这一数量未来还会大幅增加。模型的尺寸、基因序列的长度、以及所需的训练数据量,都意味着我们的确需要搭载数千颗GPU的超级计算机来完成复杂的计算。”
研究员估计,训练一个具有25亿参数的模型版本,需要约4000个GPU耗时一个多月。该团队已经在研究用于生物学的LLM,在公布论文和代码之前,他们在这个项目上已耗时约四个月。GitHub页面上有供其他研究员在Polaris和Perlmutter上运行该模型的说明。
NVIDIA BioNeMo框架可在NVIDIA NGC中心上的GPU优化软件中抢先体验。该框架将帮助研究员在多个GPU上扩展大型生物分子语言模型。作为NVIDIA Clara Discovery药物研发工具集的一部分,该框架将支持化学、蛋白质、DNA和RNA数据格式。
在SC22上莅临NVIDIA展台并观看以下特别演讲回放:
头图是研究员的LLM所测序的新冠病毒株。每个点都按新冠病毒变异株进行了颜色编码。该图由美国阿贡国家实验室的Bharat Kale、Max Zvyagin和Michael E. Papka提供。
来源:业界供稿
好文章,需要你的鼓励

英国NHS无人机快递医疗样本服务正式落地伦敦
英国国家医疗服务(NHS)正将无人机纳入常规医疗物流体系。自今年2月起,无人机每天在雷恩斯公园和圣乔治医院之间运送血液等诊断样本,飞行仅需3分钟,比公路运输快约85%,且碳排放减少高达98%。目前已有逾2000名患者受益。NHS计划将该服务扩展至圣赫利尔、克罗伊登等多家医院,最终惠及约180万名患者。该网络由英国医疗初创公司Apian与谷歌旗下Wing合作运营。

Explyt团队打造的代码智能体评测新标准:光靠“通过/失败“根本不够用
AgentLens是Explyt公司联合俄罗斯学术机构开发的AI编程助手评测基准,通过分析完整人机交互轨迹而非仅看最终结果,从五个维度评估代码智能体的真实表现。

Aetina宣布支持英伟达Jetson T3000和T2000 AI模块
边缘AI计算厂商Aetina宣布,将在其DeviceEdge AIE-KT风冷系列和新款AIE-PT无风扇平台上支持英伟达全新Jetson T3000和T2000模块。T3000基于Blackwell GPU,最高提供865 FP4 TFLOPS算力,功耗70W;T2000则提供400 FP4 TFLOPS,面向视觉AI代理和自主移动机器人等场景。两款模块预计2027年第一季度上市,支持Nemotron、Cosmos 3等英伟达AI软件生态。

机器人的“触觉觉醒“:韩国梨花女子大学如何让小型AI模型在不忘记视觉的前提下学会“感受“材质
韩国梨花女子大学提出Splash框架,通过识别AI模型中的"休眠参数"并只在其中训练触觉能力,让小型多模态AI在学会感知材质触感的同时,完整保留原有视觉语言推理能力。
2022
11/18
14:16
分享
点赞

WAIC2026 现场直击:开普勒顶流人气王,麒麟系列火爆出圈
面壁智能将密度定律带入具身智能
龙磁科技拟投3.58亿元扩建越南永磁铁氧体基地
首创一层Scale-up网络256卡全互联,摩尔线程MTT C256超节点为万卡及十万卡级集群夯实底座
从高血压诊疗入手,北京安贞医院让医疗大模型走出聊天框
西门子肖松:以场景为牵引,推动工业AI从单点实效迈向生产力跃迁
打造Token极致性价比 新华三震撼亮相2026世界人工智能大会
机器人管家系统上线!傅利叶携多款康养陪伴新品方案亮相WAIC 2026
赛那德“ 自主作业机器人天团” 登陆 WAIC:iLoabot-X+模型双升级,秀出具身场景落地硬实力
西门子Eigen工程智能体中国首发首展,荣获2026 WAIC SAIL之星奖
NVIDIA Cosmos 推动物理 AI 前沿发展
PPIO亮相WAIC 2026:发布智能模型网关,打造面向Agent时代的智能Token工厂
